
Existen múltiples causas que derivan en la aparición de una enfermedad y conocer su origen es esencial para poder tratarlas de raíz. Muchas pueden ser causadas por patógenos externos (virus, bacterias u hongos). Sin embargo, muchas otras tienen un componente genético. ¿Qué quiere decir esto? Que nuestro ADN puede presentar alteraciones que desencadenen su desarrollo.

Actualmente se ha visto que muchos genes pueden estar asociados a distintas patologías. No obstante, ¿cómo se realizan dichas asociaciones? Descubrir la implicación que puede tener la genética sigue siendo clave para mejorar el entendimiento de las enfermedades y conseguir mejores tratamientos. Con el fin de entender cómo se llevan a cabo tales asociaciones, en este artículo se mostrará la metodología que ha permitido descubrir genes involucrados en ciertas patologías.
Índice de contenido:
Tipos de mutaciones genéticas
La genética presenta una gran relevancia en la aparición de muchas enfermedades. Una gran cantidad de ellas son causadas por mutaciones en un solo gen, lo que denominamos enfermedades monogénicas. Sin embargo, gran parte de las enfermedades son enfermedades complejas, causadas tanto por factores genéticos como ambientales. En estos casos, múltiples mutaciones pueden interrelacionarse con factores ambientales (dieta, ejercicio físico, etc.) aumentando así la predisposición de su aparición.

Según su frecuencia en la población, existen distintos tipos de mutaciones que pueden contribuir a la causalidad de estas patologías. Las variantes o mutaciones comunes son aquellas mutaciones que se encuentran en la población de forma frecuente, es decir, en más de un 1% de la población; mientras que las variantes raras son las que se encuentran en menos de un 1% de la población. Actualmente sigue habiendo debate sobre qué tipo de mutación puede influir más en la patología1. Por esta razón, existe la necesidad de encontrar las causas genéticas de las enfermedades complejas, ya sea tanto de mutaciones comunes como raras.
Independientemente de la frecuencia de estas mutaciones, no todas son peligrosas para el organismo. Tal y como se explica en el artículo que lleva por título “La traducción: ¿Cómo se generan las proteínas y qué efecto pueden tener las mutaciones sobre ellas?”, algunas mutaciones pueden dar lugar a alteraciones en la estructura o síntesis de la proteína modificando su función principal. No obstante, estos cambios no tienen por qué tener siempre un efecto negativo. Por esta razón, las mutaciones pueden categorizarse de tres formas según su efecto en el individuo2:
- Mutaciones causales o de riesgo: son aquellas que tienen un efecto negativo en el individuo, ya que causan o aumentan la predisposición de padecer ciertas patologías.
- Mutaciones neutrales: son aquellas que no causan ningún efecto en el individuo.
- Mutaciones protectoras o beneficiosas: son aquellas que tienen un efecto positivo en el individuo, ya que pueden ayudar a los organismos a adaptarse al medio ambiente, protegiéndolos así de ciertas patologías o aumentando la probabilidad de supervivencia.
Estudios de asociación de genes involucrados en enfermedades
Debido a la cantidad de mutaciones que presentamos, a veces es difícil identificar cuál de todas ellas realmente está asociada a una enfermedad. Suele ser la combinación de ciertas mutaciones causales o de riesgo junto con factores ambientales lo que realmente acaban provocando una enfermedad. Por esa razón, se han desarrollado estudios de asociación de genes involucrados en patologías.
¿En qué consisten los estudios de asociación? Principalmente se basan en el uso de la epidemiología clásica en combinación con la estadística, la genética y la bioinformática con el fin de hallar las causas genéticas de una patología. Existen distintas formas de abarcar estos estudios, pero en este artículo nos centraremos en uno de los más utilizados para averiguar la asociación genética a la aparición de una patología denominado estudio de casos y controles3.
Los estudios de casos y controles están basados en dos grupos: (a) un grupo control, que consiste en un grupo de personas que no presentan la patología o la característica que se quiere estudiar y (b) un grupo de casos que son personas que presentan la enfermedad que se quiere investigar. La idea principal sería estudiar las mutaciones que contienen ambos grupos y ver si algunas de estas se encuentran más en un bando u otro. De esta forma, podríamos hallar:
- Mutaciones específicas más prevalentes en el grupo de casos y no en el de controles, lo que podría indicar una posible relación de dichas mutaciones con la patología.
- Mutaciones específicas más prevalentes en el grupo de controles y no en el de casos, indicando una posible función protectora de dicha enfermedad.
- Mutaciones específicas en ambos grupos, impidiendo observar diferencias.
A la hora de identificar estas mutaciones, existen dos métodos principales:
- A través de estudios de asociación de genoma completo (Genome-Wide Association Studies, GWAS), que permiten la identificación de mutaciones comunes.
- A través de secuenciación masiva (Next Generation Secuencing, NGS), técnica que permite detectar toda la secuencia de ADN de cada individuo y así identificar tanto mutaciones comunes como raras.
Estudio de asociación de genoma completo (GWAS)
Para estudiar la genética de enfermedades complejas en la población se desarrollaron los estudios de asociación de genoma completo (GWAS), que permiten mirar mutaciones comunes de individuos no emparentados para encontrar similitudes genéticas que les predisponen a ciertas patologías.
Identificar la secuencia completa del ADN siempre ha sido todo un reto. Por esta razón, se desarrollaron técnicas alternativas capaces de deducir el genoma completo de los individuos. Para comprender cómo se realizan estos estudios, es necesario entender cómo el material genético se hereda generación tras generación.
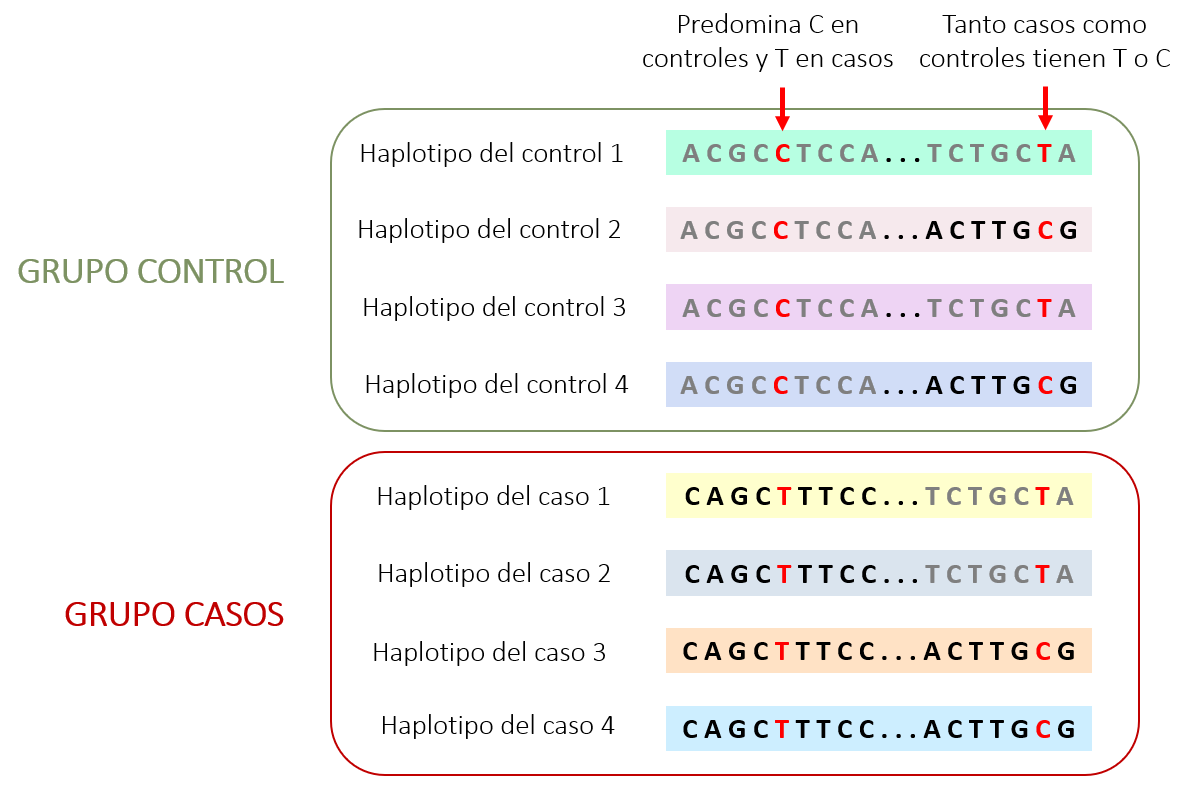
Las leyes de Mendel marcaron un antes y un después en el campo de la genética, pero no todas ellas eran completamente correctas. Según sus investigaciones realizadas en guisantes (explicadas en detalle en el artículo “¿Quién fue Mendel y cuáles fueron sus avances?”), los alelos se heredan de forma independiente, es decir, cada gen individual se transmite a la siguiente generación de forma independiente. No obstante, esto no siempre es así. Existen regiones del ADN que tienden a transmitirse juntas en forma de bloque (conocido como haplotipos), permitiendo observar toda una serie de bloques iguales en distintos individuos dentro de una misma población4.
Partiendo de este conocimiento, se inició la investigación del Proyecto HapMap con el objetivo de obtener una lista de variantes representativas de todo el genoma5. A partir del ADN de muchos individuos se pudo descubrir la presencia de variantes claves que permiten deducir el resto de la secuencia debido a que se hereda conjuntamente. Sin embargo, estas deducciones no siempre son correctas, ya que muchos individuos pueden adquirir mutaciones raras dentro de esta secuencia deducida. A pesar de ello, esta metodología resulta muy útil para aquellas mutaciones comunes entre individuos.
En resumen, los estudios GWAS se basan en identificar las mutaciones comunes en un grupo de casos (personas que padecen la patología) y controles (personas que no la padecen) para ver si se encuentran más en un grupo u otro (Figura 1). Una vez identificadas estas mutaciones, podemos ver si afectan a genes concretos, permitiendo identificar nuevos genes implicados en patologías6.
Secuenciación masiva (NGS)
Con la aparición de nuevas tecnologías capaces de secuenciar el genoma, es decir, de leer la secuencia completa del ADN, se ha facilitado la identificación de mutaciones, tanto comunes como raras, asociadas a patología. Una de las técnicas que ha permitido facilitar los estudios de asociación es la secuenciación masiva.
La secuenciación masiva permite identificar cada uno de los nucleótidos que forman el ADN de un individuo. Así, una vez secuenciado el ADN de todos los individuos de casos y controles podemos comparar diferencias entre ellos. En contraste con los estudios GWAS, que solo se observan las mutaciones comunes de cada individuo, en este caso se obtiene la información genética completa, pudiendo observar tanto las mutaciones comunes como raras.
Aunque, a priori, esta técnica parece ser mucho más interesante para detectar cualquier causa genética de una patología, el coste de secuenciar el ADN de cada individuo es mucho más elevado que realizar un GWAS. Por eso no siempre se puede recurrir a este método de estudio.
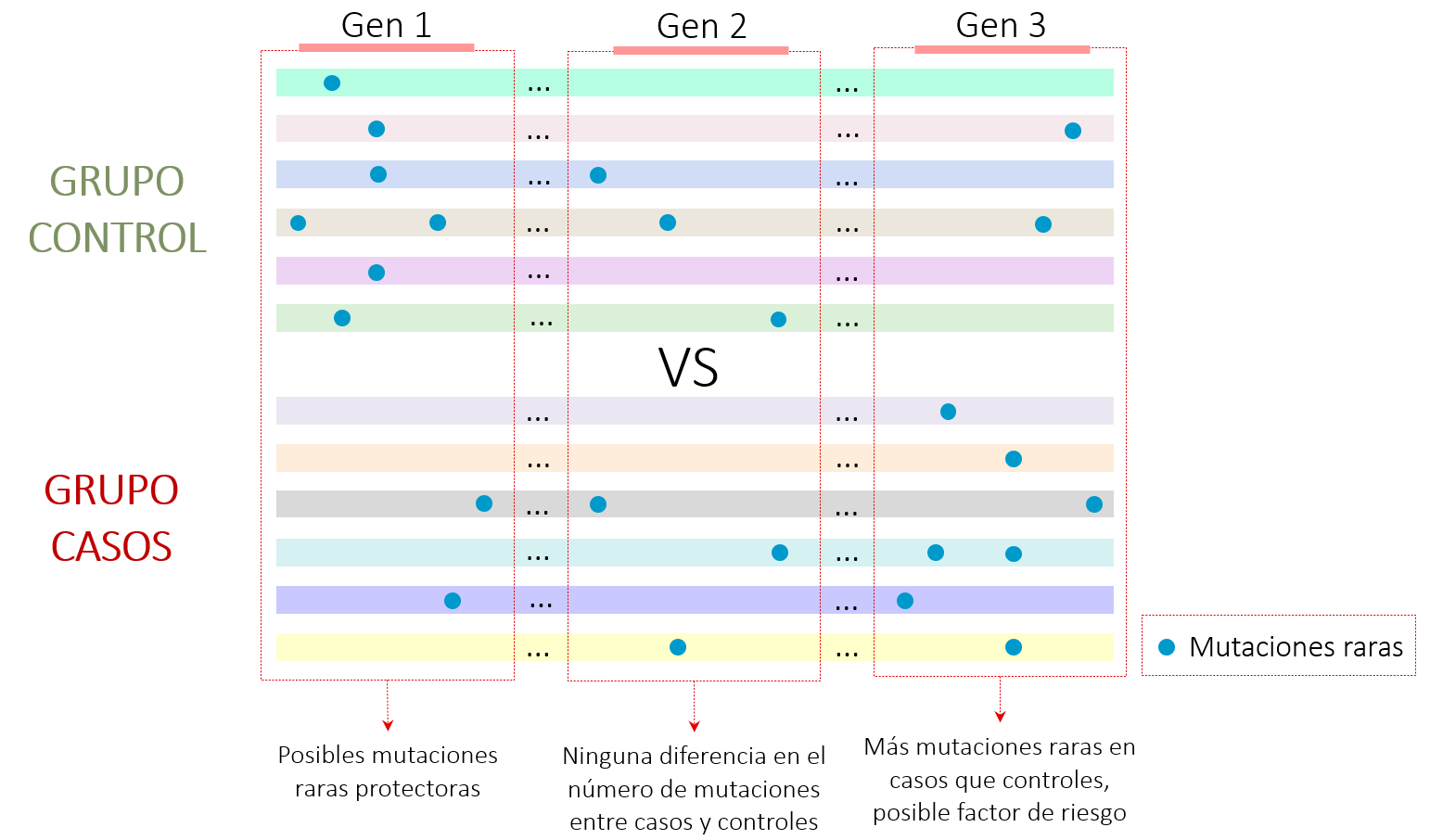
Por otro lado, investigar la importancia de las mutaciones raras también presenta algunas limitaciones. Normalmente se realizan análisis estadísticos que detectan si una mutación individual se encuentra más en un grupo u otro y, si es más abundante en casos que controles, podría indicar una asociación con la patología estudiada. No obstante, cuando hablamos de mutaciones raras, es difícil detectar personas con estas variantes, ya que se encuentran en menos de 1% de toda la población.
Para intentar solucionar este problema, algunos análisis estadísticos tienen en cuenta el conjunto de mutaciones raras que se encuentran en un gen (Figura 2). Es decir, si se observan ciertas mutaciones raras dentro de un gen prevalentes en el grupo de casos y no en el de controles podría significar que alteraciones dentro de ese gen estén causando o aumentando la predisposición de padecer dicha enfermedad7.
Validación y caracterización de las variantes
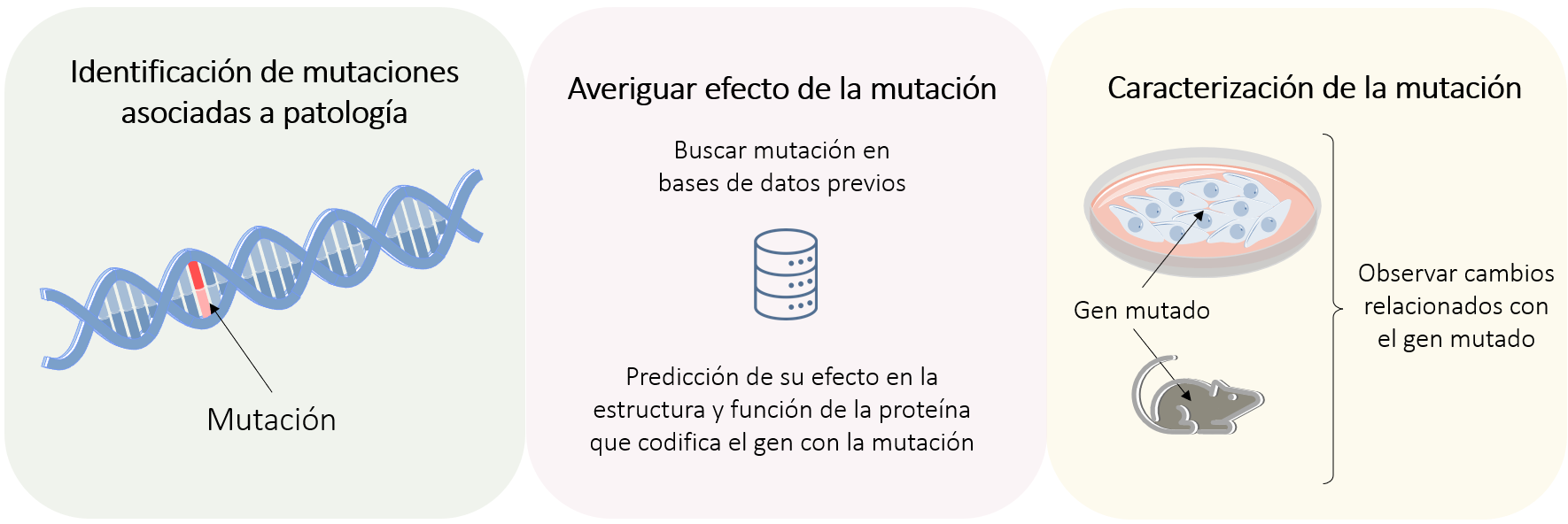
Una vez realizados estos estudios, se acaba observando la asociación de una mutación o mutaciones en genes a una patología. Sin embargo, el hecho de que ciertas mutaciones predominen en pacientes que padecen una enfermedad no significa que estén causando la patología en sí misma. Por esa razón, se ha de realizar una validación y caracterización de la mutación8. ¿En qué consiste este procedimiento? Básicamente se ha de descubrir exactamente qué está provocando esta mutación para que dé lugar a un efecto negativo o positivo en el individuo. Para ello, podemos:
- Buscar información publicada en distintas bases de datos para ver si la mutación se ha descrito antes o si se ha relacionado con alguna patología.
- Incorporar estos datos en algoritmos de predicción de efecto funcional de la proteína (son algoritmos de Machine Learning que integran el conocimiento previo de ese gen para predecir qué efecto tendrá en la estructura o síntesis de la proteína).
Independientemente de estos análisis, se deberán llevar a cabo estudios funcionales en el laboratorio capaces de dilucidar el efecto que puede tener esta mutación en el organismo. Tales estudios funcionales consisten en incorporar la mutación en un modelo biológico apropiado (células o modelos animales) para que genere esta proteína mutada (Figura 3). Posteriormente se realizarán experimentos para observar si debido a la mutación ha habido cambios en la estructura, síntesis u otros procesos moleculares y celulares en los que participa dicha proteína.
Conclusiones
Generación tras generación, se han producido mutaciones que han podido tener un efecto protector, neutro o negativo para el organismo. Con el fin de saber qué mutaciones están realmente asociadas a patología, nuevos métodos de estudios se han desarrollado en los últimos años para detectar tanto mutaciones comunes como raras. Entre ellos, encontramos los estudios de asociación de genoma completo (GWAS) capaces de relacionar mutaciones comunes a ciertas patologías y, por otro lado, estudios basados en secuenciación masiva (NGS), que permiten analizar la secuencia completa del ADN para encontrar mutaciones comunes y raras asociadas a enfermedades. Independientemente del tipo de estudio realizado, es necesaria una mejor comprensión de cómo estas variantes pueden afectar en la patología a través de la realización de estudios funcionales.
En definitiva, detectar las causas genéticas de cualquier enfermedad es primordial para entender mejor la patología y poder desarrollar nuevas terapias que consigan paliar o curar completamente dichas enfermedades.
Artículo editado por Eva Gil Hernández.
Artículos que pueden interesarte
- El genoma y los mecanismos epigenéticos
- Las mutaciones silenciosas: ¿Cómo se generan las proteínas y qué efecto pueden tener sobre ellas?
- No puedo vivir sin ti: los genes esenciales para vivir.
- ¿Podemos jugar a ser dios? La ciencia de crear vida.
Referencias
1. Schork, N. J., Murray, S. S., Frazer, K. A., & Topol, E. J. (2009). Common vs. rare allele hypotheses for complex diseases. Current opinion in genetics & development, 19(3), 212–219. doi: 10.1016/j.gde.2009.04.010
2. Mutation Effects (2021). Disponible en: https://bio.libretexts.org/@go/page/6519
3. Belbasis L., Bellou V. (2018). Introduction to Epidemiological Studies. En E. Evangelou (eds.), Genetic Epidemiology. Methods in Molecular Biology, vol 1793. Humana Press, New York, NY. https://doi.org/10.1007/978-1-4939-7868-7_1
4. Panoutsopoulou K., Wheeler E. (2018). Key Concepts in Genetic Epidemiology. En E. Evangelou (eds.), Genetic Epidemiology. Methods in Molecular Biology, vol 1793. Humana Press, New York, NY. https://doi.org/10.1007/978-1-4939-7868-7_2
5. International HapMap Consortium (2003). The International HapMap Project. Nature, 426(6968), 789–796. doi: 10.1038/nature02168
6. Dehghan A. (2018). Genome-Wide Association Studies. En E. Evangelou (eds.), Genetic Epidemiology. Methods in Molecular Biology, vol 1793. Humana Press, New York, NY. https://doi.org/10.1007/978-1-4939-7868-7_4
7. Kuchenbaecker K., Appel E.V.R. (2018). Assessing Rare Variation in Complex Traits. En E. Evangelou (eds.), Genetic Epidemiology. Methods in Molecular Biology, vol 1793. Humana Press, New York, NY. https://doi.org/10.1007/978-1-4939-7868-7_5
8. Berlanga-Taylor A.J. (2018). From Identification to Function: Current Strategies to Prioritise and Follow-Up GWAS Results. En E. Evangelou (eds.), Genetic Epidemiology. Methods in Molecular Biology, vol 1793. Humana Press, New York, NY. https://doi.org/10.1007/978-1-4939-7868-7_15





